Starting 2015 with science. This is a more in depth post about yeast phylogeny. Please have a look at the more basic post here. This time, I would like to discuss the relationship of various Brettanomyces/Dekkera strains and share my results concerning the recent WLP Brettanomyces bruxellensis Trois yeast ID crisis.
I would like to start with the Brettanomyces/Dekkera strains first. I obtained 38 Dekkera/Brettanomyces sequences (26S rDNA) from the CBS database, aligned them using MUSCLE (run in default mode, see MSA in Fig 1) and reconstructed a phylogenetic tree using MABL (model HKY85) and rendered using TreeDyn (run in default mode, Fig 2).
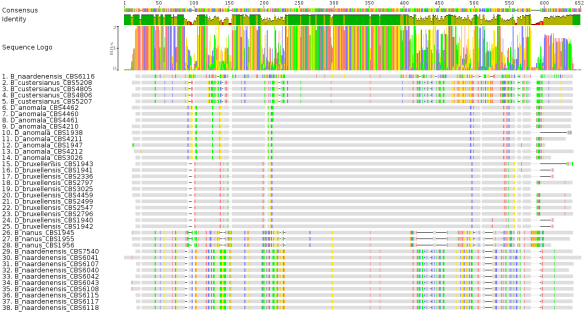
Fig 1: MUSCLE multiple sequence alignment (MSA) of 26S rDNA sequences from 38 different Brettanomyces/Dekkera strains obtained from CBS
I would like to emphasize the importance of having a look at the intermediate steps in constructing a phylogenetic tree since sequence alignments can happen in various ways. Looking at the MUSCLE output, one can already expect to see the individual strains clustered together due to their sequence similarities. However, there is one particular sequence that seems to be a bit off (sequence number 1, B. naardenensis CBS 6116). This sequence seems to be different from all the other B. naardenensis sequences shown at the bottom (sequences 29-38). Based on this result, one can expect to see CBS 6116 to be an out-group to the other B. naardenensis sequences. In summary, the alignments seem to be okay and let’s have a look at the phylogenetic tree of the 38 sequences.

Fig 2: Brettanomyces/Dekkera phylogenetic tree using 26S rDNA sequences obtained from CBS. Branch length is proportional to the number of substitutions per site (values shown in red)
As already observed in the MSA, the individual Brettanomyces/Dekkera species cluster together as expected (Fig 2). B. naardenensis CBS6116 does indeed form an out-group (isolated from lemon drink in France) and is kind of distant to the other B. naardenesis sequences. So why is this CBS 6116 sequence different?
To address this question, I tried to figure out first what other sequences are similar to CBS 6116 by BLASTing against the NCBI nr database (run in default mode). I got several hits and manually inspected the results via alignment. The CBS 6116 sequence has high sequence identities to Pichia guillermondii (Fig 3). One might reason – based on these results – that CBS 6116 is not a Dekkera/Brettanomyces strain.
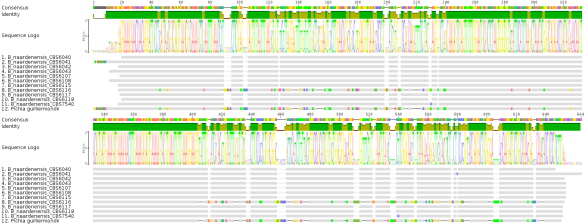
Fig 3: MSA B. naardenensis sequences & Pichia guilliermondii
Going back to the phylogenetic tree in Fig 2. It seems that all the current Dekkera/Brettanomyces species end up in the same clusters and show enough substitutions to tell individual species apart. This is very important information for everyone interested in determining the species of an unknown Dekkera/Brettanomyces strain.
WLP Brettanomyces Trois ID crisis
I got interested in the WLP story (WLP Brettanomyces bruxellensis Trois not being a Brettanoymces strain) and started by looking at the WLP Brett Trois ITS sequences I received from Omega Labs (reads 64-ITS1.ab1 and 64-ITS4.ab1). I had a quick look at the chromatograms and would not use any of the reads for my own work due to its low base quality and multiple peak calls at certain positions. Besides the read from Omega Labs, I received a read used to ID the B. Trois as Saccharoymces cerevisiae at Charles River’s Lab as well as the read published by SuiGeneris. Since I don’t have the chromatograms for these two sequences, I manually inspected the reads in various alignments to get an idea about their quality. Lets start with my analysis for the WLP Brettanoymces bruxellensis Trois ID’ing based on phylogenetic trees.
To get the most likely phylogenetic tree one has to follow some basic rules. Beginning with looking at the same shared derived homologous traits (homologous DNA sequences) and verifying that no other DNA sequence alterations impact the phylogenetic tree like sequencing errors (wrong base calls, no base call, multiple base calls etc). So far so good. I aligned some ITS2 regions from various CBS Saccharomyces strains to the various ITS2 reads from WLP’s Brettanomyces Trois (Fig 4).

Fig 4: MSA of ITS2 rDNA sequences from various Saccharomyces sp. strains obtained from CBS and ITS2 sequences from WLP B. Trois
Lets have a look at the alignment shown in Fig 4. Especially at the 64-ITS4.ab1 read from WLP B. Trois (sequence 1 shown at the top). One can easily see, that various base differences exists compared to the other sequences in the alignment. Supporting the initial idea that the DNA sequence from this read is not very reliable nor very representative (if one compares the read to the two other WLP B. Trois reads shown at the bottom). Due to the differences in this read, I would not be surprised to see this read out-grouped in the phylogeny tree later on. The two reads from Charles River’s Lab and SuiGeneris seem to be way better and similar to other Saccharomyces sp. sequences shown in the alignment.
To make the phylogeny more computational efficient and more reliable, I extracted the sequences mapping to the two WLP sequences (sequence regions from the right side in Fig 4) to receive the alignment shown in the next figure (Fig 5).

Fig 5: MSA of ITS2 rDNA sequences from various Saccharomyces sp. strains obtained from CBS (extracted)
A first look at the alignment in Fig 5 reveals some hotspots for variations like gaps and different base calls (color regions). The question I would like to address now is whether the variations are due to speciation or artifacts. Artifacts are commonly more random than variations due to speciation. A quick inspection reveals lots of random variations in the 64-ITS4 read but none/few for the two other WLP B. Trois reads.
The phylogeny tree obtained for the sequences shown in the alignment in Fig 5 is shown in Fig 6

Fig 6: Saccharomyces phylogenetic tree using ITS2 sequences obtained from CBS & WLP B. Trois from CharlesRiverLab/SuiGeneris and n64-ITS4.ab1 from Omega Lab. Branch length is proportional to the number of substitutions per site (values shown in red)
As expected the n64-ITS4 read gets out-grouped and might be interpreted as a different species. Well this is true if one just looks at the tree in Fig 6 but did not look at the previous alignments and possible reasons for the out-grouping. In this case, the quality of the base calls for the n64-ITS4.ab1 read are very poor and led to wrong base calls after all (investigated by pair-pair alignments). In summary, the differences leading to an out-grouping of WLP B. Trois based on the n64-ITS2 read are not due to the physical differences in the DNA sequence but due to sequencing errors.
In summary, the two WLP B. Trois sequences group together with other Saccharomyces cerevisiae strains. Supporting the current view that WLP B. Trois might indeed not be a Dekkera/Brettanomyces strain (at least based on ITS2 sequence homology). Or at least the samples of WLP B. Trois that float around these days.
As I do isolate and work with my own Brettanomyces strains, it happened to me various times that I was able to observe some Saccharomyces cerevisiae beside the initial Brettanomyces strain after some serial re-pitches (and I did not use Saccharomyces for the fermentation). This Saccharomyces contamination might lead to problems during the propagation. Saccharomyces cerevisiae might overgrow Brettanomyces increasing the Saccharomyces:Brettanomyces ratio even further. Eventually leading to very few Brettanomyces cells left in a population. Keeping Brettanomyces samples is not as easy as one might think. And I would not be surprised if more yeast conundrums turn up in the next years.









